
The Journey of Drug Development: From Discovery to FDA Approval
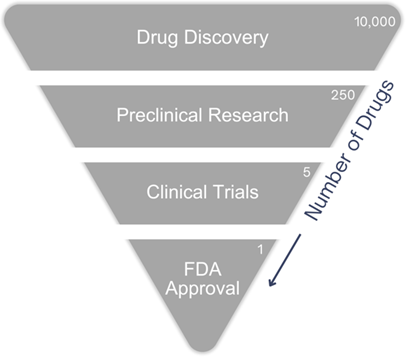
Drug development is a multifaceted and rigorous process, requiring a concerted effort from academia, pharmaceutical companies, and regulatory agencies, as well as substantial financial investment. The journey, which can often span over a decade, is marked by a series of well-defined stages, each critical to the successful launch of a new therapy. This journey begins with drug discovery and continues through preclinical research, clinical trials, and finally, regulatory approval by the FDA.
Drug Discovery: The Role of Academia and Industry
The drug development process begins with drug discovery, where the goal is to identify new compounds that have the potential to treat diseases. This stage involves a deep understanding of disease mechanisms, target identification, synthesis or discovery of compounds, and lead optimization to modulate these targets and address unmet medical needs.
A significant portion of drug discovery occurs in academic research institutions, where scientists work on understanding disease mechanisms and identifying novel biological targets. Academic research often lays the foundation for pharmaceutical innovation, providing insights that drive the early stages of drug discovery. These discoveries are then picked up by pharmaceutical companies, which have the resources and expertise to translate these early findings into potential drug candidates.
Pharmaceutical companies play a crucial role in the later stages of drug discovery, particularly in the optimization of lead compounds. They employ high-throughput screening methods, medicinal chemistry, and computational modeling to identify the most promising compounds that can modulate the biological targets identified in academic research.
Drug modalities in pharmaceutical research include small molecules, biologics, cell and gene therapies, and vaccines. Small molecules are chemical compounds, often synthesized in laboratories, while biologics include larger, complex molecules like proteins, antibodies, and gene therapies. The regulatory responsibility for these modalities falls under different FDA centers: the Center for Drug Evaluation and Research (CDER) typically oversees small molecules and some biologics, while the Center for Biologics Evaluation and Research (CBER) handles biologics, cell and gene therapies, and vaccines.
Preclinical Research: Bridging Discovery and Clinical Development
Once a potential drug candidate has been identified, it enters the preclinical research phase. This stage is crucial for evaluating the safety and efficacy of the compound before it can be tested in humans. Preclinical research involves a combination of in vitro (test tube or cell culture) and in vivo (animal) studies to gather data on pharmacokinetics (how the drug moves through the body), pharmacodynamics (the drug’s effects on the body), and toxicology.
The preclinical phase is often where the bulk of the risk in drug development lies. Only about 1 in 5,000 compounds that enter this stage will eventually become an approved drug. Preclinical research is not just about demonstrating efficacy but also about ensuring that the compound is safe for human trials. Regulatory agencies like the FDA have stringent requirements for the data generated in this phase.
A critical part of preclinical research is conducting IND-enabling studies. These studies are designed to provide the data necessary for an Investigational New Drug (IND) application, which is submitted to the FDA before beginning clinical trials in humans. IND-enabling studies typically include:
- Toxicology Studies: These studies assess the potential toxicity of the compound in one or more animal species. Acute, sub-chronic, and chronic toxicity studies are conducted to determine safe dosage levels.
- Pharmacokinetics (PK) and Pharmacodynamics (PD): PK studies determine how the drug is absorbed, distributed, metabolized, and excreted in the body. PD studies explore the drug’s biological effects and its mechanism of action.
- Genotoxicity and Carcinogenicity Studies: These studies evaluate whether the compound has the potential to cause genetic mutations or cancer. These studies are typically required if there are concerns based on the drug’s structure or class.
- Reproductive Toxicology: These studies assess the potential impact of the drug on fertility and embryonic development.
- Safety Pharmacology Studies: These studies assess the potential effects on major organ systems, such as the cardiovascular and respiratory systems.
Clinical Trials
The clinical trial process is divided into several stages, each serving a critical function:
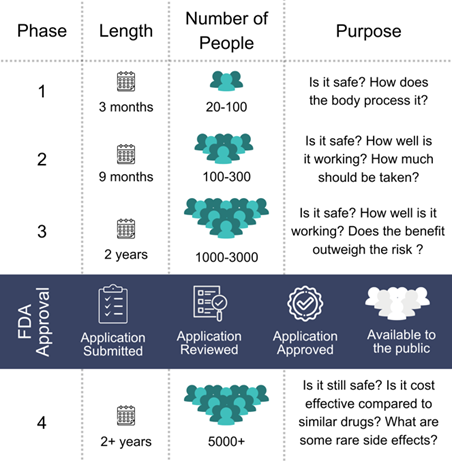
- Phase 1: In the first phase of clinical trials, the primary focus is on safety. A small group of 20 to 100 healthy volunteers or patients, depending on the nature of the drug, is administered the drug to determine its safety profile. This includes understanding how the drug is absorbed, distributed, metabolized, and excreted (ADME) in the body. The main objectives are to identify any potential side effects, the maximum tolerated dose, and pharmacokinetic properties. This phase typically lasts several months and helps establish a foundation for further studies by ensuring that the drug does not pose significant safety risks.
- Phase 2 Trials: After Phase 1 confirms safety, Phase 2 trials expand the focus to the drug’s efficacy. This phase involves a larger group of patients, typically between 100 to 300, who have the condition the drug is intended to treat. These trials last several months to two years and are designed to evaluate the drug’s effectiveness, determine the optimal dose, and continue safety assessments. Phase 2 trials are critical for identifying the most promising candidates for further testing, as they provide the first indication of the drug’s therapeutic potential.
- Phase 3 Trials: Phase 3 trials are pivotal studies that involve large-scale testing of the drug in diverse populations, often ranging from 1,000 to 3,000 patients or more. These trials compare the new drug to existing standard treatments or a placebo to determine its effectiveness and monitor side effects. Conducted over several years, Phase 3 trials provide the comprehensive data required for regulatory approval, including statistical evidence of the drug’s safety and efficacy. This phase is crucial for confirming the drug’s benefits and ensuring it is safe for widespread use in the general population.
FDA Review and Approval
After successful completion of clinical trials, the drug sponsor submits a New Drug Application (NDA) or Biologics License Application (BLA) to the FDA. This application includes all data from the preclinical and clinical studies, information about the drug’s composition and manufacturing, and the proposed labeling.
The FDA review process can take several months to years. The agency assesses whether the drug’s benefits outweigh its risks and whether it meets the FDA’s rigorous standards for safety, efficacy, and quality. In some cases, drugs that address unmet medical needs may qualify for priority review or accelerated approval pathways. If approved, the drug can be marketed and made available to patients.
Post-Market Surveillance
After a drug is approved by regulatory authorities such as the FDA, Phase 4 trials, also known as post-marketing surveillance studies, are conducted to monitor its long-term safety and effectiveness. These trials involve thousands of patients and are designed to detect any rare or long-term adverse effects that may not have been apparent in earlier trials. Phase 4 studies are essential for ensuring the drug continues to be safe and effective in real-world use. Additionally, these studies can lead to updates in dosing recommendations, safety warnings, or even the drug’s withdrawal from the market if necessary. In some cases, Phase 4 trials may also uncover new therapeutic uses for the drug.
Drug Modalities and Their Implications
Different drug modalities, such as small molecules, biologics, gene therapies, and cell therapies, each have unique challenges and requirements throughout the drug development process. For example, small molecules often have the advantage of oral administration and simpler manufacturing processes, but they may face challenges in specificity and off-target effects. Biologics and gene therapies, on the other hand, are highly specific and can target complex diseases, but they require more complex manufacturing and delivery methods, such as intravenous injections, as well as presenting significant regulatory, ethical, and technical challenges. Additionally, biologics and gene therapies often require cold chain logistics (temperature-controlled supply chains) due to their sensitivity.
Conclusions
The drug development process is a monumental endeavor, involving the integration of scientific discovery, rigorous testing, and regulatory scrutiny. Despite the challenges and high costs, the process is essential for bringing new, life-saving therapies to patients. The evolution of drug modalities, from small molecules to gene therapies, continues to push the boundaries of what is possible in medicine, offering hope for treatments that were once unimaginable.
The journey from discovery to market is long and fraught with challenges, but it is also one of the most rewarding paths in science, offering the possibility of making a profound impact on human health.


